Sindromul Aicardi
Autor: Redacția ROmedic
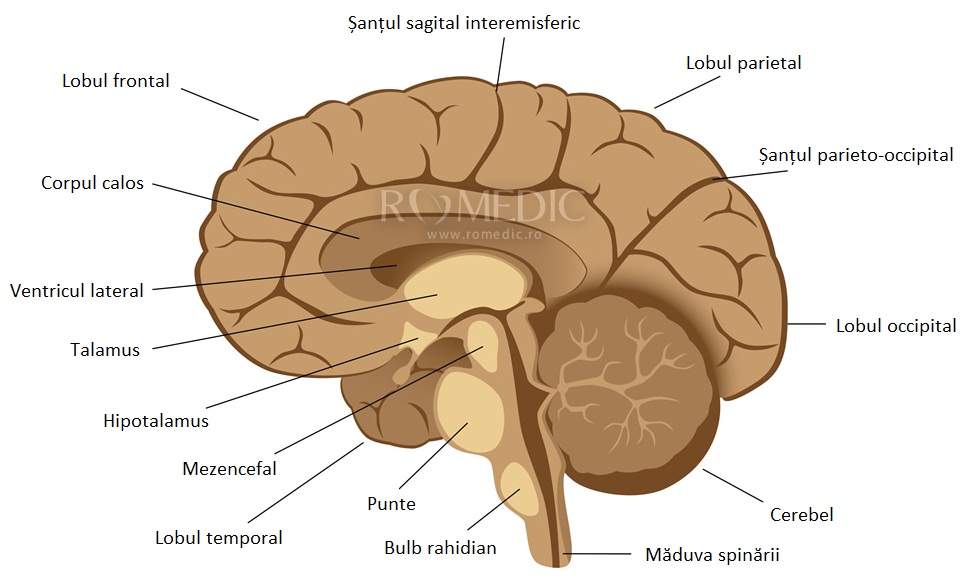
Sindromul Aicardi se refera la o boala genetica in care factorul mutagen actioneaza exclusiv asupra cromozomului dominant X. In nomenclatura afectiunii se regaseste numele neurologului francez Dr. Jean Aicardi, care a descris pentru prima data aceasta boala extrem de rara a copilariei.
Dintre cazurile raportate (doar 500 pe plan mondial), majoritatea sunt fetite intre 3 si 5 luni. Acest sindrom congenital poate sa apara extrem de rar si la baietii cu mutatia genetica Klinefelter (structura cromozomiala XXY).
Problema centrala este lipsa partiala sau totala a corpului calos intre cele doua emisfere cerebrale, care afecteaza grav comunicarea intracerebrala. Pana in prezent nu s-a descoperit nici un tratament, iar speranta de viata este foarte scazuta.
Etiologie
Factorul declansator al sindromului il reprezinta o mutatie genetica care afecteaza exlusiv cromozomul X. Se presupune ca aceasta mutatie apare in saptamanile 4-8 de sarcina, in perioada embriogenezei, printr-o distensie exagerata a tubului neural. Gena mutagena dominanta va determina patologia.
Boala este genetica, dar nu ereditara, fiind vorba de o mutatie care apare pentru prima data. Baietii care au un defect cromozomial pe gena X nu pot supravietui.
Sindromul nu poate sa apara la baieti in lipsa unei mutatii cromozomiale preexistente. De exemplu, in cazul sindromului Klinefelter, baiatul are doi cromozomi X si unul Y. Daca unul din cromozomii X este atacat de factorul mutagen Aicardi, boala se va manifesta si la baieti.
- Traducere engleza: Aicardi Syndrome
- Traducere germana: Aicardi-Syndrom
Manifestari clinice
Mutatia genetica din sindromul Aicardi determina:
- malformatii cerebrale
- imunosupresie
- malformatii ale sistemului nervos
- calcifierea radacinilor nervoase
- ajungandu-se pana la atrofie cerebrala.
Clinic apar manifestari patologice la nivel:
ocular:
- Leziuni oculare caracteristice (defecte albe sau galbene in jurul papilei optice), cu lipsa de pigment, ceea ce le diferentiaza de keratitele si retinitele infectioase;
Microftalmie
Cataracta
Dezlipire de retina
Coloboma
craniocerebral:
- Microcefalie
- Asimetrie hemifaciala
- Microftalmie
- Fanta labiopalatina (buza-de-iepure)
musculo-scheletal:
- Malformatii ale scheletului
- Hemivertebre
- Vertebre sudate
- Anomalii costale
- Scolioza
neuronal
- Retard mintal sever
- Chist porencefalic
- Crize epileptice
- Dezvoltare motorie deficitara (datorita hipotoniei, spasmelor, sau hemiplegiei)
Diagnostic
De cele mai multe ori diagnosticarea sindromului Aicardi se face intamplator, cu ocazia unui examen oftalmologic sau datorita crizelor infantile cu aspect epileptic.
Examenul de certitudine este cel imagistic (CT sau RMN), care arata calcifierea radacinilor nervoase sau chiar atrofie cerebrala.
Relevante pot si crampele musculare specifice crizelor comitiale.
Analiza lichidului cefalorahidian indica cresterea numarului de leucocite.
La indicatia medicului se poate face si o electroencefalografie, pentru masurarea activitatii electrice a creierului si identificarea tipului de crize comitiale.
Tratament
Vindecarea sindromului Aicardi nu este in prezent posibila. Geneticienii continua insa cautarea cauzei si a unui leac pentru aceasta patologie.
Tratamentul simptomatic se bazeaza pe:
- anticonvulsivante: pentru ameliorarea crampelor musculare
- ACTH (hormonul adrenocorticotrop): pentru ameliorarea crizelor comitiale care nu raspund la anticonvulsivantele uzuale;
- fizioterapie :pentru incetinirea degradarii motorii;
Prognostic
Prognosticul sindromului Aicardi este trist, cu degradarea progresiva si ireversibila a functiei locomotorie si vizuala (pana la orbire). Majoritatea pacientilor mor in prima decada de viata, rareori supravietuiesc pana in a doua sau a treia, avand nevoie de ingrijire medicala continua.
- Helicobacter pylori
- Colagenoza nespecifica. Polimiozita
- Ganglioni axilari inflamati
- Urticarie colinergica
- Boala Behcet
- Hiv sau paranoia?
- Alergie la toti alergenii posibili
- Ce vaccin sau tratament pot face sa-mi intaresc imunitatea
- Exista vreo sansa sa NU am lupus...?
- Ganglioni limfatici inflamati de doi ani de zile